
A Not So Simple $12 Donation
Last Friday was a special little moment for me. I got to make a small donation of $12 to Charity: Water. While a donation to this organization is not anything new to me, since I give monthly as being one of the first Spring Members at Charity: Water, but what makes this donation notable is where that money came from.
That small $12 has been part of a year-long experiment I’ve been testing on myself, where I’ve been trying to utilize the power of investment growth to generate exponentially larger donations.
On March 26th, 2018, I opened an investing account with Betterment and made an automatic investment of $20 every month. The goal was to capitalize on market growth through a diversified portfolio that Betterment provides, and every year from here on out, I’ll liquidate 5% of the account balance and donate that.
Why did I put $240 ($20 x 12 months) into an investing account, and only donate $12 instead of just donating the whole $240? Because I’m playing the long game. It is part of a larger theory I’ve working for the past year in what I’ve been calling “Perpetual Value” which is that if you continually add to an account that is growing in value, you can pull of small portions of the fund to fuel other initiatives while the growth rate in the fund will replenish the balance, making the fund not only self-sustaining, but growing exponentially as well. You can read this concept more in depth here: https://grantxstorer.com/microfoundations/
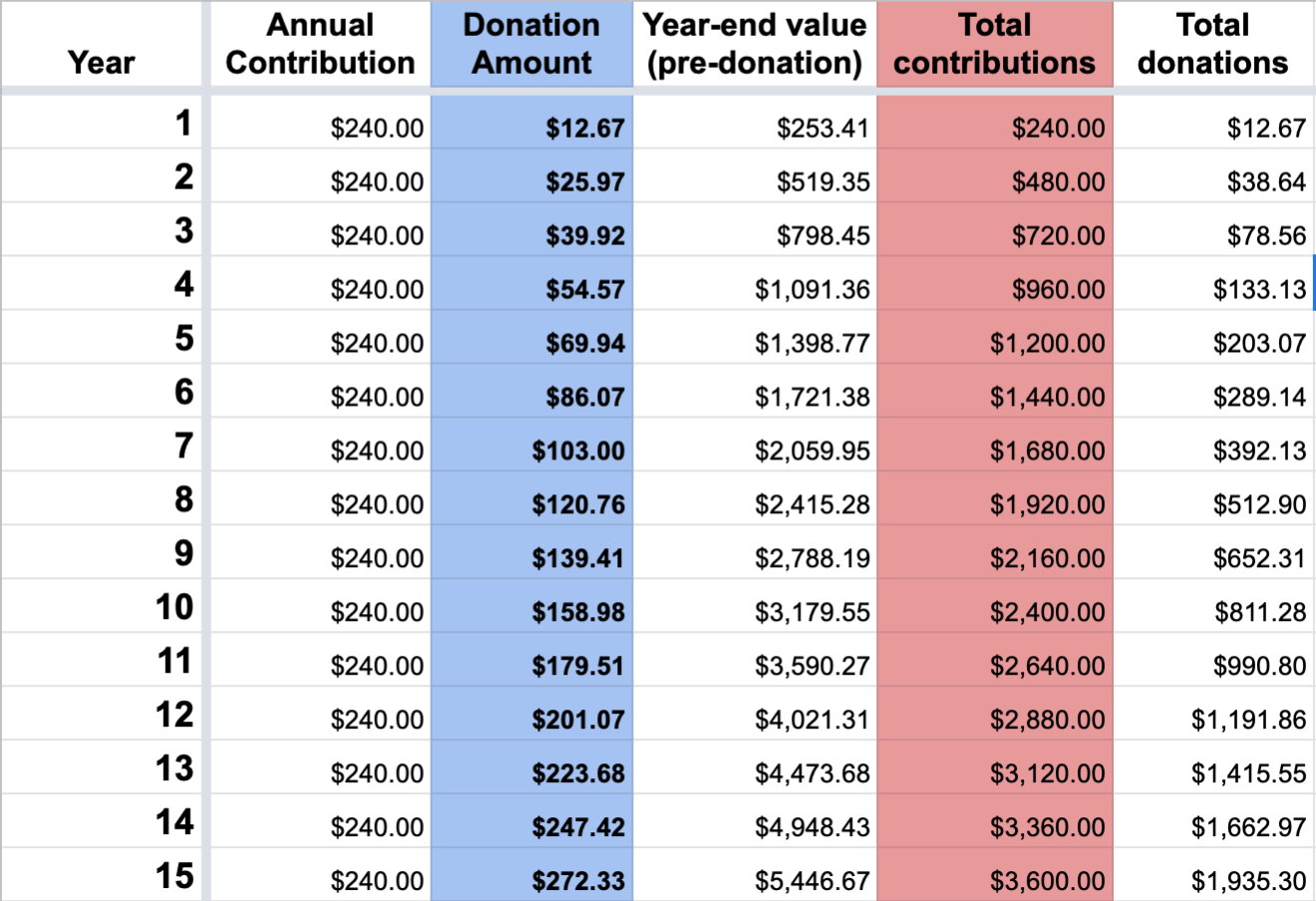
To keep things simple, just look at what happens in a simplified model where I decide to give $20 per month every month over the next 15 years. Assuming I could generate a 10% stable growth over this time, by the time we get to year 15, I would have contributed $3,600 of my own money to the investment account. The interesting thing however, is look at the total donations at the bottom-right of the table, it says to-date I would have donated $1,935.30. On top of that, the fund balance I have is $5,174.34. This means that I’ve already given over half of all the money I invested into this account, and yet, the account balance is 140% the size of what I’ve invested.
From here on out, every year I will get to donate to whoever I choose for more money that I’ll actually put into the investment account that year. If I still was just putting in $240 per year into the account, the amount I actually donate is going to be increasingly more than $240 (if we expand the model to year 50, I’d be donating $2600 that year, which is over 1,000% what I would be putting in for that year!).

One Year Later
That was the theory. I wrote all this out as a projection a year ago, and while it made sense conceptually, I needed to start putting it into practice to see how well reality matches up with theory.
According to my model, after one year, I would have an account balance of $253.41, which would allow me to make a $12.67 donation. In reality, I’m currently behind with a balance of $244.94 which would permit me to make a $12.25 donation.
I know, these numbers are too small to make any real difference right now, but this is just the first step into the process, and it’s cool to actually get some measurable data back.
What’s Being Donated?
Since the numbers are too small, I just simply rounded down to $12 for my first donation, but here’s a quick analysis of what was actually being donated here.
The fund grew by $4.94 over the past year, so how much of that $12 came from growth, and how much came from principle? To find this out we just need to find the relative percentage that principle and growth have on the total value. I won’t bore you with the math here, but what this results in that of the $12 donated, $11.76 was from principle, while $0.24 came from growth. While again it isn’t much, it does show that I was able to preserve 2% of what I invested, and that 2% of what I donated came from market growth.
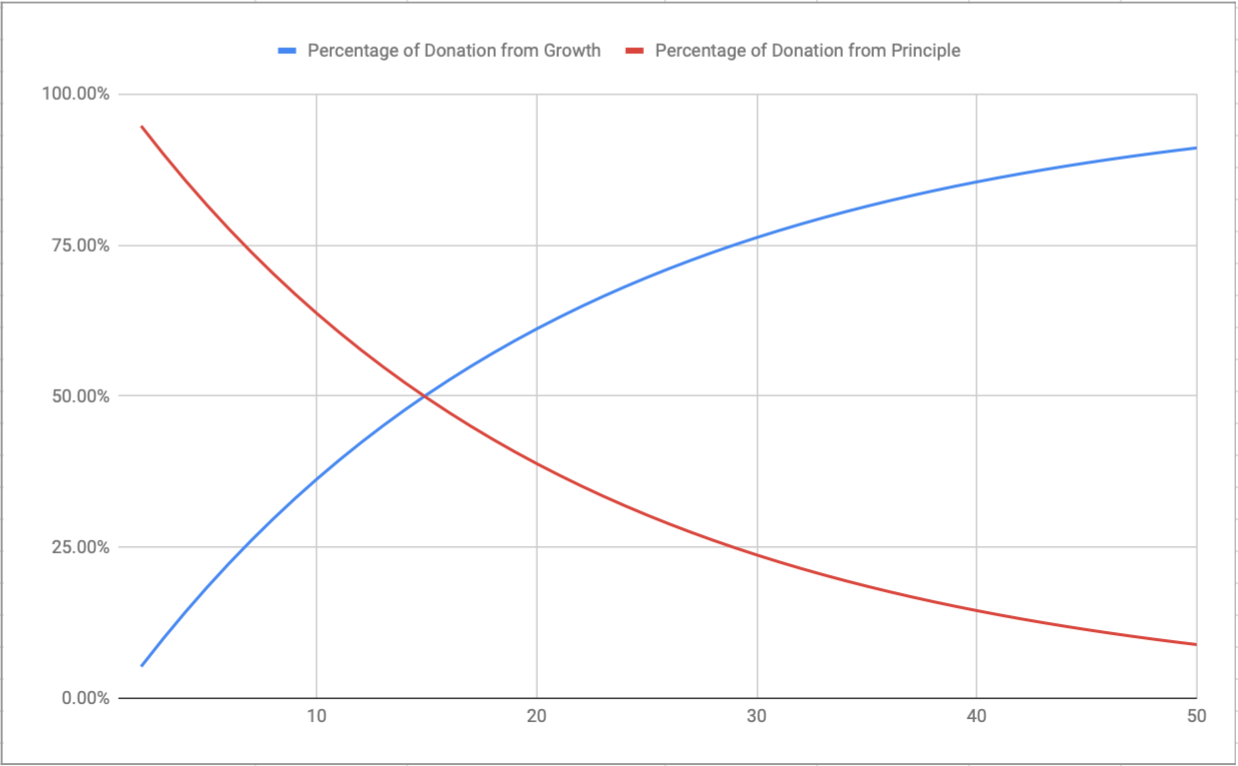
As time progresses, the distribution of what is being donated should increasingly drift towards investment growth over principle. The more this progression takes place, the more principle is preserved to produce more growth, which in turn results in larger donations in the future.
This graph demonstrates how investment growth slowly progresses to become the primary source of a donation over the 50 year period:
It’s a tiny start, but it’s exciting to emerge from the theoretical and into the real world. I made a $12 donation today, but if there is validity to this model, I can risk putting more of my resources dedicated to impact investing through this model, which would further expand my capacity to make a positive impact.


